The neonate with a
neuromuscular disorder
Eugenio Mercuri and Francesco Muntoni
Chapter Contents
- Introduction
- Obstetric history and clinical assessment
- Congenital muscular dystrophies
- Congenital myotonic dystrophy
- Congenital myopathies
- Motoneuron disorders
- Mitochondrial encephalomyopathies
- Summary
- Acknowledgments
- References
Introduction
Neuromuscular disorders may present with hypotonia, weakness and/or contractures in the newborn period10. However, these may also be the presenting signs in infants with abnormalities of the central nervous system. With the easier access to brain MRI in the neonatal period, it is possible to identify those infants whose hypotonia is related to congenital or acquired brain lesions and those infants in whom there is both central and peripheral nervous system involvement. In many cases it is possible to differentiate infants with a primary neuromuscular disorder from those with involvement of the CNS by detailing the obstetric history, by careful observation and by performing a careful physical examination10, 12. In this manner, only selected infants will need further investigation with imaging techniques.
< prev | top | contents | next >
Obstetric history and clinical assessment
Reduced fetal movements, breech presentation and polyhydramnios are frequently observed in children with weakness with onset in utero. Contractures, skin dimpling and poor dermatoglyphic patterns are all indicators of poor fetal movements and can provide information on the severity and timing of onset of the immobility. These signs are highly suggestive of a ‘peripheral’ lesion and with the exception of a few conditions are rarely associated with CNS involvement. The exceptions include those conditions that affect the fetus in the first months of pregnancy, such as neuronal migration disorders (e.g. bilateral opercular syndrome), or antenatal basal ganglia injury from a severe hypoxic–ischemic insult (e.g. attempted maternal suicide)22.
The presence or absence of antigravity movements is also important. These have to be observed when the infant is at the peak of the activity, such as when crying or when in discomfort. While infants with neuromuscular disorders will show little change in their movement pattern in response to pain or when crying, children with CNS, metabolic or syndromic disorders may have a severely floppy posture which is interrupted by occasional antigravity movements. The distribution of wasting, weakness and hypotonia is also important, but might differ in individual conditions and only rarely can provide a diagnostic clue.
Reflexes are also an important part of the examination. Normal reflexes in a floppy infant almost exclude a severe peripheral neuropathy or motor neuron disorder and make a severe myopathy unlikely.
Other signs, such as an abnormal pattern of respiratory muscle involvement, major bulbar weakness with inability to suck and to clear secretions are frequently observed in children with some of the congenital myopathies, but can also be a feature of ‘central’ involvement, as observed in the infants with basal ganglia or brain stem lesions (see Chapter 6).
In this chapter we focus on the involvement of the central nervous system in infants with conditions that affect primarily the skeletal muscle or the motoneuron, or in diseases in which both central and peripheral nervous system are involved as part of more general multisystemic involvement.
< prev | top | contents | next >
Congenital muscular dystrophies
The term ‘congenital muscular dystrophies’ includes a group of disorders which share several clinical and pathological features such as contractures, weakness and hypotonia at birth or in the following few months and a dystrophic pattern on muscle biopsy. The spectrum of clinical signs in the neonatal period is variable, and in some of the congenital muscular dystrophy syndromes involvement of the CNS can dominate the clinical picture.
Three well-defined syndromes, Fukuyama congenital muscular dystrophy, Walker–Warburg syndrome, muscle–eye–brain disease, which are all associated with structural brain changes, were separated from a ‘pure’, or classical form of congenital muscular dystrophy, not associated with structural brain changes by an International Consortium in 199311, 13. Imaging studies, however, have demonstrated that a proportion of children with ‘pure’ congenital muscular dystrophy showed white matter changes, and these changes were consistently related to the deficiency of the laminin α2 chain of merosin, an extracellular matrix protein. In contrast, children with normal expression of merosin were initially described as having a normal MRI30. More recently, however, there have been a few descriptions of other brain changes, such as cortical dysplasia and cerebellar hypoplasia, not only in the merosin-deficient group but also in the children with normal merosin, suggesting that the involvement of the brain in congenital muscular dystrophy is more complex than initially assumed47. Table 14.1 illustrates the current but ever growing classification of congenital muscular dystrophy based on clinical and MRI findings, and when known, on protein and molecular data.
Table 14.1 Classification of the congenital muscular dystrophies
| Form | MRI findings | Protein | Molecular data |
|---|---|---|---|
| Fukuyama CMD | Mycropolymicrogyria, pachygyria, cerebellar involvement, abnormal signal in white matter | Fukutin | 9q31 |
| Walker–Warburg syndrome | Type II lyssencephaly, hydrocephalus, brainstem and cerebellar hypoplasia | ||
| Muscle–eye–brain disease | Pachgyria and polymicrogiria Brain stem and cerebellar hypoplasia, periventricular white matter frequent but not constantly observed |
1p | |
| CMD with primary merosin-deficiency Typical form With cortical dysplasia With cerebellar hypoplasia |
Diffuse white matter changes WM changes and cortical dysplasia WM changes and cerebellar hypoplasia |
Laminin α2 (merosin) | 6q2 |
| CMD with secondary merosin-deficiency With muscle hypertrophy, rigidity of the spine With severe phenotype and normal brain MRI With microcephaly and mental retardation With microcephaly, and cerebellar hypoplasia With mental retardation and cerebellar cysts |
Normal Normal Normal Cerebellar hypoplasia Cerebellar cysts |
1q-42 | |
| Merosin positive CMD Typical form + Mental retardation + Cerebellar hypoplasia |
|||
| CMD, congenital muscular dystrophy; WM, white matter. | |||
< prev | top | contents | next >
FUKUYAMA CONGENITAL MUSCULAR DYSTROPHY
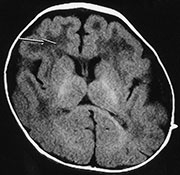
Fig. 14.1 Fukuyama muscular dystrophy (severe). Infant aged 7 months with the severe form of Fukuyama muscular dystrophy. The severe type makes up approximately 10% of all cases. The MRI appearances are more marked with more prominent white matter changes (arrow) that are clearly seen on the T1 weighted (SE 450/15) sequence. This infant also had a right-sided micro-ophthalmia and retinal detachment (not shown). (With permission, Professor Yukio Fukuyama.)
This form of muscular dystrophy was first described by Fukuyama et al. in 1960 and is almost exclusively confined to Japan18. The inheritance is autosomal recessive and the locus has been assigned to chromosome 9. Recently, the gene responsible for this form has been identified and the protein product named fukutin. The clinical features of the Fukuyama congenital muscular dystrophy are early onset with mild to moderate hypotonia at birth and a progressive course with more increasing weakness, joint contractures, marked elevation of creatine kinase, moderate to severe mental retardation and frequent association with epilepsy. Ocular abnormalities, such as myopia and optic nerve atrophy occur in approximately 70% of these children but are rarely severe.
A severe form has been demonstrated in approximately 10% of cases (Fig. 14.1).
Brain MRI
MRI shows structural changes consisting of pachygyria and polymicrogyria and abnormal signal intensity within the white matter (Fig. 14.2). Sequential MRI studies have demonstrated that the abnormal white matter appearance is associated with delayed myelination rather than with dysmyelination or demyelination as the abnormal signal intensity improves with age. In fact, in these children myelination, though very delayed, still occurs, following the patterns normally observed at earlier ages in normal children. The frontal lobes are the last areas in which the abnormal signal persists. Ventricular dilatation has also been frequently recorded. Cerebellar hypoplasia may be seen but is not frequent28.
")
")
Fig. 14.2 Fukuyama muscular dystrophy. Infant aged 9 months of age. T1 weighted spin echo (SE 450/15) (a) and T2 weighted fast spin echo (FSE4000/100) (b) sequences. There is abnormal signal intensity within the white matter. This is most marked in the frontal lobes (long arrow) and is more clearly seen on the T2 weighted images. The appearance of the frontal cortex is consistent with polymicrogyria (short arrow). This is also most clearly seen on the T2 weighted images. (With permission, Professor Yukio Fukuyama.)
Pathology
The abnormal cortex shows localized areas of pachygyria and polymicrogyria with markedly irregular arrangements of neurons. Areas of normal cortex can be observed. Recent studies in fetuses have suggested that a defect of the basement membrane might play a role in the genesis of the brain lesions38.
WALKER–WARBURG SYNDROME
This is an autosomal recessive disorder characterized by the combination of muscular, brain and ocular involvement. The clinical picture can be very variable and signs of CNS involvement predominate7–9. Although milder phenotypes have been described, these infants generally show severe hypotonia, poor visual attention and decreased alertness. Muscle weakness in the first year of life can be mild. Serum creatine kinase can also be normal in the initial phases of the disorder or only mildly elevated, but tends to increase with age.
Muscle pathological studies show a dystrophic pattern but the changes may, however, be less severe with only minimal variation in fiber size in the first months of life.
Ocular abnormalities consist of retinal dysgenesis in all cases but other abnormalities such as microphthalmia, cataracts, optic nerve hypoplasia and anterior chamber malformations have also been described.
Brain MRI
Brain MRI shows a type II lissencephaly with the typical micropolygyric cobblestone cortex. Cerebellar hypoplasia, mainly affecting the vermis, is also a constant feature. White matter is also severely abnormal showing dysmyelination or cystic changes. Other features, such as brain stem hypoplasia, Dandy–Walker syndrome or encephaloceles have been described, in various combinations.
Pathology
The cobblestone cortex consists of cortical and leptomeningeal abnormalities. These abnormalities are more severe than the ones observed in Fukuyama and muscle– eye–brain disease. Some children have widespread agyria, whereas other show a combination of pachygyria and polymicrogyria with only small areas of agyria. The whole brain can be covered by glial tissue and the cortex completely disorganized with no horizontal layers.
CASE 14.1 WALKER–WARBURG SYNDROME
Perinatal history
This Indian male infant was born at 34 weeks’ gestation by cesarian section. Early antenatal ultrasound scan detected cerebral ventricular dilatation. At birth the infant showed some dysmorphic features, such as torricephaly, small jaw and low set ears. He had poor visual alertness and did not feed well.
Clinical course
At 2 weeks a ventriculoperitoneal shunt was inserted because of an increasing head circumference. At 5 weeks he still showed generalized hypotonia and muscle weakness, more severe in the proximal distribution associated with mild antigravity strength in the limbs. The pattern of movements was very poor. There was some visual fixation but no tracking. Sucking movements were present but there was poor stripping.
Serum creatine kinase were markedly elevated (5620IU).
At 3 months he showed a variable tone and better antigravity power in the limbs despite the persistent proximal weakness. Visual alertness was still very reduced but sucking had improved. Ophthalmology review demonstrated lamellar cataracts but no retinal abnormalities.
A muscle biopsy showed some variability in fiber size.
MRI
Brain MRI at 5 weeks (Fig. 14.3) demonstrated marked ventricular dilatation with a thickened featureless cortex, consistent with type II lissencephaly. There was also hypoplasia of the brain stem and cerebellar hemispheres. The basal ganglia were rudimentary, the lateral and third ventricle were grossly enlarged.
")
")
")
")
Fig. 14.3 Walker–Warburg syndrome: case 14.1. Imaged at 5 weeks of age. Inversion recovery (IR 3800/30/950) sequence (a) and T2 weighted spin echo (SE 2700/120) sequence (b). Transverse plane. There is gross ventricular dilation with a smooth thick cortical rim (arrow). T1 weighted spin echo (TE 860/20) in sagittal (c) and coronal (d) planes. There is cerebellar hypoplasia (short arrow) and rudimentary basal ganglia (long arrow).
MUSCLE–EYE–BRAIN DISEASE
Muscle–eye–brain disease is an autosomal recessive form of congenital muscular dystrophy originally described by Santavuori in 1977. It is relatively frequent in Finland and has been recently mapped to chromosome 1p.
Clinical signs are usually present at birth or in the first months of life. Late presentation is less common. Ocular abnormalities consist of myopia, retinal defects and abnormal electroretinogram. Some of the ocular symptoms can become evident only after the first years of life. These children show severe mental retardation and, often, epilepsy35.
Brain MRI
Brain MRI shows extensive abnormalities of neuronal migration, such as pachygyria and polymicrogyria. Brain stem and cerebellar hypoplasia are also frequent but not constantly observed. The white matter can show areas of abnormal signal but these are not very prominent and, when present, are localized in the periventricular areas.
Pathology
Neuropathological studies have shown coarse gyri with a nodular appearance of the surface and agyric areas. Microscopically, the cortex appeared disorganized without horizontal lamination. The cerebellar cortex is also disorganized.
MEROSIN-NEGATIVE CONGENITAL MUSCULAR DYSTROPHY
In 1994 Tome first reported that a proportion of children with pure congenital muscular dystrophy showed a deficiency of the laminin α2 chain (merosin), an extracellular membrane protein normally present in skeletal muscle44. The laminin α2 chain gene has been mapped to chromosome 6q22–2347. Children with merosin deficiency have, in general, a more severe clinical course than the children with congenital muscular dystrophy but normal merosin. They are usually symptomatic at birth or in the first few weeks of life with hypotonia and muscle weakness, a weak cry and, in 10–30% of cases, with contractures. Motor nerve conduction velocity is generally reduced36.
White matter changes on MRI are a constant feature in these children. The changes diffusely involve the hemispheres, sparing the corpus callosum and the brain stem. The degree of involvement can be variable between individuals but is consistent within siblings29, 30. The changes may be difficult to identify within the first months of life (Fig. 14.4 and Fig. 14.7) when it can be difficult to differentiate between unmyelinated white matter and abnormal myelination but become more evident around 6 months and, by the age of 12 months the pattern is similar to that observed in older children24, 25(Fig. 14.7) Despite their dramatic appearance on imaging, these changes do not result in significant functional impairment in these children and cognitive abilities are usually within normal limits. Visual and somatosensory evoked potentials, in contrast, are generally delayed or absent23. The only other sign of the involvement of the central nervous system is epilepsy, which has been observed in 10–30% of children47.
The significance of the white matter changes is not clear as laminin α2 expression appears to be limited to the blood–brain barrier45. A pathological study has suggested that the changes observed on MRI might be the result of an increase in water content in the brain15. This would explain why the changes seem so striking on imaging yet do not give a significant functional impairment.
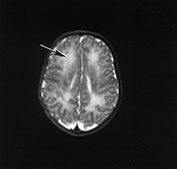
Fig. 14.4 Merosin-negative congenital muscular dystrophy. Male infant born at term with marked hypotonia and weakness. He had mild contractures. His creatine kinase was markedly elevated. Imaged at 5 days of age. T2 weighted fast spin echo (FSE 300/120) sequence. There is abnormal high signal intensity within the periventricular white matter (arrow).
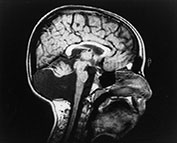
Fig. 14.5 Merosin-negative congenital muscular dystrophy. Male child aged 9 years. T1 weighted spin echo (SE 860/20) sequence in the sagittal plane. There is marked hypoplasia of the cerebellar vermis (arrow) and the cerebellar hemispheres (not shown).
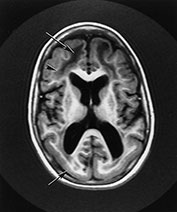
Fig. 14.6 Merosin-negative congenital muscular dystrophy: case 14.2. Male child aged 11 years. Inversion recovery (IR 3400/30/800) sequence. There is abnormal low signal intensity within the white matter (long arrow). There is preservation of the normal signal form myelin within the U fibers (arrowhead). The cortex of the occipital and temporal lobes is lissencephalic (short arrow).
Recent studies have demonstrated, however, that besides the diffuse white matter changes, which have been observed in all the patients with merosin-deficient congenital muscular dystrophy, other patterns of brain lesions may also be observed. The association of white matter changes and cerebellar hypoplasia has now been described by several authors. The hypoplasia generally involves the vermis and is not associated with severe mental retardation (Fig. 14.5)25, 42, 47. In contrast, the association of white matter changes and cortical dysplasia is normally associated with severe mental retardation and epilepsy (Fig. 14.6)25, 32, 37, 47 although children with normal mental function have been reported4.
CASE 14.3 MEROSIN-NEGATIVE CONGENITAL MUSCULAR DYSTROPHY
Perinatal
This caucasian female infant was born at 42 weeks’ gestation by forceps for failure to progress. Birth weight was 3.410kg. She was noted to be floppy soon after birth and transferred to the neonatal unit on day 1 because of hypoglycemia, slow feeding and difficulty in maintaining body temperature.
At 3 weeks she still showed generalized hypotonia with marked head lag, no effort against gravity in ventral suspension and mild equino-varus talipes deformities. Some antigravity movements were present in both arms and legs but the pattern of spontaneous movements was very poor.
Investigations
Creatine kinase was grossly elevated at 4000IU/l (normal = <200IU/l). Electromyography of the quadriceps was interpreted as normal. A needle muscle biopsy of the quadriceps at 5 weeks of age showed dystrophic changes. On immunocytochemistry, the laminin α2 chain of merosin was deficient but not entirely absent, confirming a diagnosis of merosin-deficient congenital muscular dystrophy.
Clinical development
The child is now 3 years old. She has remained floppy and her motor milestones were all delayed. At 3 years she is able to sit without support and to stand with support. Intellectual development is appropriate for her age. Her head circumference is above the 90th centile.
Magnetic resonance imaging
MRI was done at 3 weeks and 6, 12 and 17 months (Fig. 14.7).
")
")
")
")
")
")
")
")
Fig. 14.7 Merosin-negative congenital muscular dystrophy: case 14.3. Female child aged 3 weeks. Inversion recovery (IR 3800/30/950) (a) and T2 weighted spin echo (SE 2700/120) (b) sequences showed the appropriate pattern for her age. Myelin was present in the posterior limb (arrow) of the internal capsule, brain stem, pons and cerebellum. Areas of unmyelinated white matter had abnormal low signal intensity on IR but were unremarkable on T2 weighted images. There were small cysts in the left lentiform nucleus and mild dilatation of the left lateral ventricle were seen (c,d,e). Aged 6 months. Diffuse abnormalities of both myelinated and unmyelinated white matter were present. These were seen as low signal intensity on the inversion recovery (IR 3400/30/800) sequence (c) with corresponding high signal on T2 weighted spin echo (SE 2700/120) (d). Myelin was present in the anterior and posterior parts of the corpus callosum, anterior and posterior limbs of the internal capsule (e). The ventricles were mildly dilated (f,g,h). Aged 12 months. Both inversion recovery (IR 3400/30/800) and T2 weighted spin echo (SE 2700/120) images showed abnormal signal intensity throughout the periventricular and subcortical white matter. Myelin was present in the internal capsule, brain stem and cerebellum. Abnormal signal intensity was also seen in the centrum semiovale and small amounts of myelinated white matter were present in the occipital lobes. At 17 months, there was no discernible change from the scan performed at 12 months.
Congenital muscular dystrophy with secondary merosin-deficiency
In the last few years there have been several description of other forms of congenital muscular dystrophy with secondary merosin-deficiency which do not link to the LAMA 2 gene. Brain MRI plays an important role in the differential diagnosis of these forms. Although this is a heterogeneous group, none of these cases reported so far has shown the typical white matter changes invariably observed in patients with primary deficiency of merosin. Brain MRI is also helpful in the characterisation of the phenotype in the different forms which have recently been reported.
Congenital muscular dystrophy with secondary merosin deficiency, muscle hypetrophy and rigidity of the spine linked to chromosome 1q42
This form, characterized by proximal girdle weakness, generalized muscle hypertrophy, rigidity of the spine and contractrures of the tendo Achilles, was originally described in a consanguineous family from the United Arab Emirates and has recently been assigned to chromosome 1q42. Brain MRI is normal5.
Other forms of secondary merosin deficiency have been described in which all the known loci of CMD were executed by linkage analysis. The variability in clinical phenotype suggest genetic heterogeneity.
Congenital muscular dystrophy with secondary merosin deficiency and normal brain MRI
We have recently described a form of CMD in which the clinical phenotype strongly resembles that observed in the 6q-merosin deficient form with the exception of the brain MRI which is completely normal26.
Congenital muscular dystrophy with secondary merosin deficiency, microcephaly and normal structural brain
Another form of CMD with secondary merosin deficiency and normal brain MRI has been reported in 2 siblings by Topaloglu et al41. At variance with the previous cases however, these siblings have microcephaly and mental retardation.
Congenital muscular dystrophy with secondary merosin deficiency, microcephaly-muscle hypertrophy-cerebellar hypoplasia
Villanova et al have recently reported 5 patients from 4 Italian families with a severe phenotype characterised by severed hypotonia, weakness and joint contractures since birth, muscle hypertrophy and severe mental retardation46. Brain MRI showed an abnormal posterior cranial fossa with enlargement of the cisterna magna, variable hypoplasia of the vermis of cerebellum and periventricular white matter changes.
Congenital muscular dystrophy with secondary merosin deficiency and cerebellar cysts
A consanguineous Turkish family with this phenotype was recently reported39. Characteristic features were early-onset hypotonia, generalized muscle wasting, weakness, join contractures and moderate mental retardation. Brain MRI revealed multiple small cysts in the cerebellum, without cerebral cortical dysplasia or white matter changes.
MEROSIN-POSITIVE CONGENITAL MUSCULAR DYSTROPHY
The diagnosis of merosin-positive congenital muscular dystrophy is usually made after having excluded all the remaining congenital muscular dystrophy syndromes. The clinical picture of children with merosin-positive congenital muscular dystrophy is very heterogeneous. Classically, this form has been described as having normal MRI, but recent studies have shown that a small proportion of these children show some brain changes, ranging from non-specific localized white matter changes, possibly secondary to antenatal or perinatal insults, to the presence of structural abnormalities, such as cerebellar hypoplasia17,31, 43.
Recently, the locus for a form of merosin-positive congenital muscular dystrophy characterized by marked rigidity of the spine and respiratory failure in the first or second decade of life has been assigned to chromosome 1p.
CASE 14.4 MEROSIN-POSITIVE CONGENITAL MUSCULAR DYSTROPHY
Perinatal
This Asian female infant was born at 26 weeks’ gestation by emergency cesarian section for fetal distress. Her birth weight was 3.240kg. She required ventilation and was transferred to the neonatal unit. On examination she presented dysmorphic features, was floppy and showed a limited range of movements, slow feeding and reduced visual alertness. She also had contractures of her knees, elbows and neck.
Investigations
Creatine kinase was grossly elevated at 4000IU/l (normal = <200IU/l). Electromyography of the quadriceps was suggestive of a myopathic process. A needle muscle biopsy of the quadriceps at 5 days of age showed dystrophic changes. On immunocytochemistry, the laminin α2 chain of merosin was present, confirming a diagnosis of merosin-positive congenital muscular dystrophy.
Clinical development
The child showed a progressive improvement. She was weaned off the ventilator after the neonatal period and feeding was established soon after. Her tone and her weakness have also improved. She is now 4 years old and she is able to walk and almost to run.
Magnetic resonance imaging
MRI of the brain was performed at 2 days and repeated at 16 months (Fig. 14.8).
")
")
Fig. 14.8 Merosin-positive congenital muscular dystrophy: case 14.4. Inversion recovery (IR 3800/30/950) sequence (a). Infant born at 36 weeks’ gestation and imaged at 2 days of age. There is a widened extracerebral space frontally. The cortical folding of the frontal lobes is slightly immature. There is a small amount of high SI consistent with early myelination in the posterior limb of the internal capsule (arrow) (a) T1 weighted spin echo (SE 860/20) aged 16 months. The frontal space is still markedly widened (b). Myelination has progressed appropriately.
< prev | top | contents | next >
Congenital myotonic dystrophy
The main presenting features of congenital myotonic dystrophy in the newborn are generalized hypotonia, contractures and marked difficulty in sucking and swallowing, usually requiring tube feeding. Respiratory difficulties are also frequent and often responsible for neonatal death34. These infants show a striking facial diplegia with a triangular-shaped open mouth and are unable to close their eyes completely. Skeletal deformities, such as talipes, are very common. Unlike the adult form, in the neonatal period there is no evidence of myotonia and the EMG is usually normal. The typical myotonic discharges only appear later on in life. The muscle pathology may be confusing as this can show either minimal non-specific changes or features suggestive of myotubular myopathy.
A good clinical and antenatal history and a careful examination of the mother provide the diagnostic clue. A history of reduced fetal movements in pregnancy and polyhydramnios can be elicited in most instances. The demonstration of grip or percussion myotonia and facial weakness in the mother establishes the diagnosis of myotonic dystrophy.
The severe neonatal weakness involving the respiratory and swallowing muscles shows a gradual improvement tending to resolve after the first months of life. Very severely affected neonates can, however, require ventilator support for weeks and some may never be weaned off the ventilator. Although the brain is structurally normal in these children, non-specific abnormalities are often observed (Fig. 14.9). The incidence of ventricular dilatation in these infants has been reported to be between 70 and 80%. Other abnormalities, such as intraventricular hemorrhage and periventricular leukomalacia have also been reported19, 40.
")
")
")
Fig. 14.9 Congenital myotonic dystrophy: case 14.5. Female infant born at 36 weeks’ gestation. Her mother was diagnosed as having myotonic dystrophy after the birth of a previously affected child. This pregnancy was complicated by polyhydramnios. The infant was born by emergency cesarian section with Apgar scores of only 3 at 1min. She was intubated for lack of respiratory effort and remained ventilator dependent for 41 days. She was imaged at 6 weeks of age. T1 weighted (SE 860/20) (a,b) and T2 weighted (SE 2700/120) (c) spin echo sequences. There is mild ventricular dilation. There are additional small probably hemorrhagic lesions seen as high signal intensity on T1 weighted images (long arrow). These are not easily visible on the T2 weighted images (c). Myelination within the posterior limb of the internal capsule is appropriate for age (a,b).
Hashimoto et al. have reviewed the MRI in seven cases with congenital myotonic dystrophy showing ventricular dilatation in all, associated with periventricular or deep white matter changes (6/7), cortical atrophy (3/7), small corpus callosum (4/7) and small brain stem (2/7)19.
< prev | top | contents | next >
Congenital myopathies
Involvement of the CNS may also be observed in some of the congenital myopathies, such as the severe variant of nemaline myopathy and X-linked myotubular myopathy.
NEMALINE MYOPATHY
This myopathy is characterized by the presence of rods on muscle biopsy. The classical congenital form, observed in early infancy, is characterized by mild floppiness and swallowing problems, and is not usually associated with CNS involvement. In contrast, the more severe and usually fatal congenital form characterized by arthrogryposis and ventilator dependency at birth, is more frequently associated with brain involvement, such as mild dilatation of the ventricles or cortical atrophy. Structural brain abnormalities are, however, not observed in this form.
CASE 14.6 NEMALINE MYOPATHY
Perinatal
This African male infant was born at 34 weeks’ gestation by emergency cesarian section for breech presentation. His birth weight was 2.360kg. He was intubated soon after birth showing very poor respiratory movements. On examination he showed severe hypotonia, with absence of spontaneous movements, mild contractures in the wrist, reduced visual alertness and poor sucking.
Investigations
His creatine kinase was normal (123U/l). A needle muscle biopsy of the quadriceps at 5 days of age showed the classical features of nemaline myopathy. Cranial ultrasound showed marked posterior echo densities and diffuse echo densities over the cerebellum. He died at 4 months of age.
Magnetic resonance imaging
Brain MRI was performed at 10 days of age (Fig. 14.10).
")
")
")
Fig. 14.10 Nemaline myopathy: case 14.6. T1 weighted (SE 860/20) (a,b) and T2 weighted (SE 2700/120). There is a large subdural hemorrhage (long arrow). Multiple small areas (short arrows) of high signal intensity on T1 weighted images (a,b) and low signal intensity on T2 weighted images (c) are consistent with hemorrhagic lesions.
X-LINKED MYOTUBULAR MYOPATHY
Children affected with X-linked myotubular myopathy have a very similar clinical picture to the severe congenital form of myotonic dystrophy, with the additional involvement of the extraocular muscles, giving rise to ophthalmoplegia. The long-term survival in this form is extremely poor because of the severity of the respiratory and bulbar muscle involvement.
CASE 14.7 X-LINKED MYOTUBULAR MYOPATHY
Perinatal
This African male infant was born at 35 weeks’ gestation by cesarian section for fetal distress. Early antenatal ultrasound scan detected polyhydramnios. The mother also felt reduced fetal movements. At birth the infant showed no respiratory effort and was intubated. On examination he showed ophthalmoplegia, facial weakness, severe hypotonia, no antigravity movements, and no visual alertness. Sucking and rooting were also absent.
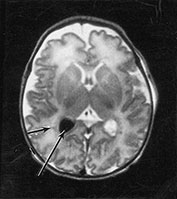
Fig. 14.11 X-linked myotubular myopathy: case 14.7. T1 weighted spin echo (SE 860/20) sequences. Infant aged 10 days. There is an intraventricular hemorrhage (long arrow) and small parenchymal hemorrhagic lesions in the white matter (short arrow). The parenchymal hemorrhagic lesions are easier to see using a T1 weighted sequence.
Investigations
Creatine kinase was grossly elevated at 4000IU/l (normal = <200IU/l). Electromyography of the quadriceps was suggestive of a myopathic process. A needle muscle biopsy of the quadriceps at 5 days of age showed dystrophic changes. Cranial ultrasound was normal.
Magnetic resonance imaging
Brain MRI showed diffuse abnormal signal intensity in white matter with small petecchial foci consistent with hemorrhage. There was a widened extracerebral space (Fig. 14.11).
Clinical development
At 5 months the infant is still oxygen dependent, is still very floppy and has shown only a minimal improvement with onset of some antigravity movements. His alertness is also very poor with no auditory orientation and only occasional opening of the eyes.
< prev | top | contents | next >
Motoneuron disorders
Spinal muscular atrophy (SMA) type 1 (Werdnig–Hoffman disease) is the most common disease of the motoneuron in the newborn. The condition characteristically involves the proximal limb and the intercostal respiratory muscles more severely, while it spares the facial muscles and the diaphragm; clinically evident bulbar involvement is frequent but relatively mild. Infants affected by this form usually die within the first year, or before 18 months of age. The recent discovery of the gene defect for SMA (deletion of the telomeric copy of the SMN (survival motor neuron) provides a very useful and rapid diagnostic tool as a deletion of this gene can be found in more than 98% of children with SMA 1. Although in Werdnig–Hoffman disease there is usually no associated central nervous system involvement, non-specific changes such as cerebral atrophy or other ischemic changes have been observed in some of these children, mainly associated with prematurity or birth asphyxia33.
PONTOCEREBELLAR HYPOPLASIA TYPE I
Unlike Werdnig–Hoffman disease, clinical and radiological evidence of CNS involvement is a major feature of the form of spinal muscular atrophy associated with pontocerebellar hypoplasia type I3. Studies have shown that this form is not allelic to SMA14 and the gene responsible for this form has not yet been identified.
Affected infants show a combination of clinical signs related to both motoneuron disorder and central nervous system involvement. The clinical course is progressive and respiratory and feeding difficulties, already present at birth or in early infancy, become more severe.
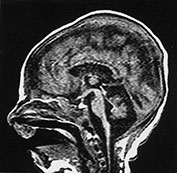
Fig. 14.12 Pontocerebellar hypoplasia type 1: case 14.8. Female infant born at 32 weeks’ gestation and imaged at 3 weeks of age. T1 weighted spin echo (SE 860/20) sequence. There is hypoplasia of the cerebellum, medulla and mid-brain.
The MRI shows hypoplasia of the cerebellar vermis and, often also of the cerebellar hemispheres, associated with a thin brain stem and pons (Fig. 14.12). Pathological studies have shown normal brain weight with extreme hypoplasia of pons and cerebellum. Cerebellar folia were poorly developed while vermal folia were relatively well preserved with general loss of granular cells and atrophy of dentate and inferior olivary nuclei. A near total loss of nuclei was also observed in the pons with reactive gliosis3.
The differential diagnosis is with other forms, which show cerebellar and brain stem hypoplasia and peripheral nerve involvement, such as the carbohydrate-deficient glycoconjugate syndromes.
CASE 14.8 PONTOCEREBELLAR HYPOPLASIA TYPE 1
Perinatal
This female infant was the first child born to healthy non-consanguineous parents of Asian origin. At 28 weeks’ gestation polyhydramnios was noted and at 32 weeks there was poor fetal growth and movement. End diastolic flow velocities were absent. The mother was given dexamethasone and the baby was delivered by cesarian section. The infant had poor respiratory effort requiring immediate intubation and ventilation; her birth weight was on the third centile. She had widespread contractures involving large and small joints. Her posture was characterized by hypotonia, internal rotation of arms and ulna deviation of fingers with adducted thumbs in clenched fists. There was virtually no spontaneous movement, even after painful stimuli. The baby was unable to swallow.
Clinical development
She never opened her eyes spontaneously and remained ventilator dependent. At 1 month of age she became progressively more difficult to ventilate, with sepsis and increasing acidosis. Intensive care was withdrawn and the child died soon afterwards.
Magnetic resonance imaging
Brain MRI revealed a large subcerebellar space with symmetrical cerebellar and mesencephalon hypoplasia. The mid-brain also appeared small and the ventricular system was dilated (Fig. 14.12).
Only a limited postmortem examination was allowed and this revealed generalized muscle atrophy, affecting the distal muscles more severely. The brain was small and immature with a hypoplastic cerebellum and moderate dilatation of the lateral ventricles. The spinal cord was grossly normal.
PERIPHERAL NERVE DISORDERS ASSOCIATED WITH CNS INVOLVEMENT
Several metabolic diseases can affect both the peripheral nerves and brain but in the majority of them the onset occurs after the neonatal period. A possible underlying metabolic disorder, however, must always be considered when investigating a severely ill and floppy newborn. These infants often present with respiratory or cardiac problems and an abnormal neurological examination with reduced tone and a reduced level of alertness or seizures. A clinical examination can provide some useful additional information such as the presence of dysmorphic features or visceromegaly which can aid the differential diagnosis.
The carbohydrate-deficient glycoconjugate syndromes deserve a particular mention, because of structural changes observed in the cerebellum and in the brain stem. These syndromes include a group of genetic disorders characterized by a deficiency of the carbohydrate moiety of glycoconjugates20, 21. Type I is the most common and has been related to a deficit in phosphomannomutase. Infants present with dysmorphic features, hypotonia, and hyporeflexia, abnormal eye movements and poor feeding. The clinical presentation, however, can be variable, even within siblings and other signs in the neonatal period are skeletal abnormalities, hepatomegaly, proteinuria and cardiomyopathy. Retinitis pigmentosa, joint contractures, epilepsy and stroke-like episodes are frequent but usually occur at a later stage. The involvement of the peripheral nerves is shown by abnormal motor and sensory nerve conduction velocity.
Serial MRI is useful in differential diagnosis, showing brain stem and cerebellar hypoplasia20, 21.
The diagnosis is made by isoelectrofocusing and immunofixation of serum transferrin and can be confirmed by finding decreased phosphomannomutase in fibroblasts or leukocytes.
Other conditions, such as Refsum’s disease and some peroxisomal disorders can show similar signs, with dysmorphic features, slow nerve conduction and global delay. In some of these such as neonatal adrenoleukodystrophy (Fig. 14.13) or Refsum’s, the involvement of the brain may not be easily detectable in the neonatal period when it is difficult to differentiate between the unmyelinated white matter and signs of dysmyelination.
")
")
Fig. 14.13 Neonatal adrenoleukodystrophy. Female infant born at 38 weeks’ gestation with dysmorphic features, hypotonia and contractures at the knee. She developed neonatal seizures. Imaging was performed at 5 weeks of age. T1 weighted spin echo (SE 860/20) sequence (a). There is minimal high signal intensity from myelin within the internal capsule (short arrow). The level of myelination is at least 1 month behind. The white matter has a very low signal intensity (long arrows). T2 weighted spin echo (SE 2700/120) sequence (b). The white matter has a very high signal intensity. There is no obvious signal from myelin within the posterior limb of the internal capsule (arrow).
< prev | top | contents | next >
Mitochondrial encephalomyopathies
Mitochondrial disorders are described separately from the other metabolic disorders because of the possible association of brain changes with peripheral nerve and skeletal and cardiac muscle involvement. Only a few of the mitochondrial disorders affecting children and adults have neonatal onset (see chapter 17). Lactic acidosis is the landmark of most of these forms which, in the neonatal period, are mainly related to defects of the respiratory chain and, in particular to deficiency of complex I and III and, more commonly, of complex IV. There have been several descriptions of a fatal infantile form with cardiac and renal involvment, seizures, hypotonia and respiratory distress. In these cases CT and MRI generally show cortical and subcortical atrophy and dysmyelination1 (Fig. 14.14). The muscle can be normal, or reveal lipid–glycogen storage.
")
")
")
")
")
")
")
")
Fig. 14.14 Mitochondrial myopathy. Male infant born at 42 weeks’ gestation presenting with an initial diagnosis of hypoxic–ischemic encephalopathy. He was noted to be profoundly hypotonic and his seizures persisted. Magnetic resonance proton spectroscopy showed elevated lactate throughout the brain but much more marked in the white matter on proton spectroscopy. He was imaged at 3 weeks, 4 months and 16 months.
Images at 3 weeks. Inversion recovery (IR 3800/30/950) sequence (a) T2 weighted spin echo (SE 2700/120) sequence (b,c). There is a rather thin signal from myelin within the internal capsule (long arrow) for 45 weeks. There are areas of increased signal intensity within the white matter (short arrows). Images at 16 months (d,e,f,g,h) show marked atrophy of the white matter with decreased myelination. The corpus callosum is present and shows some myelination but is very thin (short arrow) (h). The cerebellum has become atrophied (long arrow) (h).
A neonatal onset has also been described for the Leigh syndrome or subacute necrotizing encephalopathy6. The underlying enzyme defect can only be detected in 20–25% of the cases and is often related to complex IV or I deficiency and, in some cases to pyruvate dehydrogenase deficiency. The clinical manifestations of Leigh syndrome are very variable and often the overt symptoms appear, even in the infantile form, after a relatively symptom-free period. Clinical signs consist of hypotonia and developmental regression with onset of movement disorders and seizures. In the neonatal forms the symptoms are evident soon after birth with absence of poor acquisition of new milestones.
The MRI in Leigh syndrome shows a typical pattern with abnormal signal in the bilateral lenticular nuclei and in the caudate with subsequent cavitation. Cerebellar hypoplasia is also frequent. MRI spectroscopy shows high levels of lactate in the basal ganglia, even in the absence of increased serum lactate. Muscle biopsy may be morphologically normal or show only non-specific changes but show biochemical abnormalities. Nerve conduction velocity (NCV) can be abnormal.
Summary
- MR brain imaging will help in correctly diagnosing infants with signs of neuromuscular disease.
- MRI is identifying an increasing range of associated brain abnormalities within the congenital muscular dystrophies.
- Follow-up imaging in infants with neuromuscular disorders may show progressive changes within the white matter.
- MR spectroscopy may be useful for differentiating metabolic disease from primary neuromuscular disorders.
Acknowledgments
We thank Mary Rutherford for preparing all the images and legends.
< prev | top | contents | next >
References
- Aicardi J (1998) Diseases of the Nervous System in Childhood. London, MacKeith Press.
- Akaboshi S, Ohno K and Takeshita K (1995) Neuroradiological findings in the carbohydrate-deficient glycoprotein syndrome. Neuroradiology 37, 491–495.
- Barth PG (1993) Pontocerebellar hypoplasia. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev 15, 411–422.
- Brett FM, Costigan D, Farrell MA et al. (1998) Merosin-deficient congenital muscular dystrophy and cortical dysplasia. Eur J Paed Neurol 2, 77–82.
- Brockington M, Sewry CA, Herrmann R, et al. (2000). Assignment of a form of congenital muscular dystrophy with secondary merosin deficiency to chromosome 1q42. A J Hum Gen, 66, 428–435.
- Cocker SB and Thomas C (1995) Connatal Leigh disease. Clin Pediatr 34, 349–352.
- Dobyns WB and Truwitt CL (1995) Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics 26, 132–147.
- Dobyns WB, Kirkpatrick JB, Hittner HM et al. (1985) Syndromes with lyssencephaly II. Walker–Warburg and cerebro-oculo-muscular syndromes and a new syndrome with type II lyssencephaly. Am J Med Genet 22, 157–195.
- Dobyns WB, Pagon RA, Armstrong D et al. (1989) Diagnostic criteria for Walker–Warburg syndrome. Am J Med Genet 32, 195–210.
- Dubowitz V (1969) The floppy infant. Clinics in Developmental Medicine 31, London, Spastics International/Heinemann.
- Dubowitz V (1994) Workshop report on 22nd ENMC-sponsored meeting on congenital muscular dystrophy held in Baarn, The Netherlands, May 14–16 1993. Neuromusc Disord 4, 75–81.
- Dubowitz V (1995) Muscle Disorders in Childhood, 2nd edn. London, Saunders.
- Dubowitz V, Fardeau M (1995) Workshop report on 27th ENMC-sponsored meeting on congenital muscular dystrophy held in Baarn, The Netherlands, April 22–24 1994. Neuromusc Disord 4, 253–258.
- Dubowitz V, Daniels RJ and Davies KE (1995) Olivopontocerebellar hypoplasia with anterior horn cell involvement (SMA) does not localize to chromosome 5q. Neuromusc Disord 5, 25–29.
- Echenne B, Pages M and Marty Double C (1984) Congenital muscular dystrophy with cerebral white matter spongiosis. Brain Dev 6, 491–495.
- Echenne B, Rivier F, Jellali AJ et al. (1997a) Merosin positive congenital muscular dystrophy with mental deficiency, epilepsy and MRI changes in the cerebral white matter. Neuromusc Disord 7, 187–190.
- Echenne B, Rivier F, Tardieu M et al. (1997b) Congenitalmuscular dystrophy and cerebellar atrophy. Neurology (1998)50, 1477-80.
- Fukuyama Y, Kawazura M and Haruna H (1960) A peculiar form of congenital muscular dystrophy. Paediatr Univ Tokyo 4, 5–8.
- Hashimoto T, Tayama M, Myazaki M et al. (1995) Neuroimaging study of myotonic dystrophy I. Magnetic resonance imaging of the brain. Brain Dev 17, 24–27.
- Jaeken J and Casaer P (1997) Carbohydrate-deficient glyconjugate (CDG) syndromes: a new chapter of neuropaediatrics. Eur J Paed Neurol 2/3, 61–66.
- Jaeken J, Stibler H and Hagberg B (1991) The carbohydrate-deficient glycoprotein syndrome: a new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand Suppl 375, (monograph).
- Maalouf E, Battin M, Counsell S et al. (1997) Arthrogryposis multiplex congenita and bilateral mid-brain infarction following maternal overdose. Eur J Paed Neurol 5/6, 183–186.
- Mercuri E, Muntoni F, Berardinelli A et al. (1995) Somatosensory and visual evoked potentials in congenital muscular dystrophy: correlation with MRI changes and muscle merosin status. Neuropediatrics 26, 3–7.
- Mercuri E, Pennock J, Goodwin F et al. (1996) Sequential study of central and peripheral nervous system involvement in an infant with merosin-deficient CMD. Neuromusc Disord 6, 425–429.
- Mercuri E, Gruter-Andrew J, Philpot J et al. (1999) Cognitive abilities in children with congenital muscular dystrophy: correlation with brain MRI and merosin status. Neuromuse Disord 9, 383–387.
- Mercuri E, Sewry CA, Brown SC, et al. (2000). Congenital muscular dystrophy with secondary merosin deficiency and normal brain MRI: a novel entity? Neuropediatrics 31:186–189.
- Mercuri E, Rutherford M, De Vile C, et al. (2001) Early white matter changes on brain magnetic resonance imaging in a newborn affected by merosin-deficiency congenital muscular dystrophy. Neuromusc Disord, 11:297-9.
- Osawa M, Sumida S, Suzuki N et al. (1997) Fukuyama type congenital progressive muscular dystrophy. In: Fukyama Y, Osawa M and Saito K (Eds) Congenital Muscular Dystrophies. Amsterdam, Elsevier Science.
- Philpot J, Sewry C, Pennock J et al. (1995) Clinical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromusc Disord 5, 301–305.
- Philpot J, Topaloglu H, Pennock J et al. (1995) Familial concordance of brain magnetic resonance imaging changes in congenital muscular dystrophy. Neuromusc Disord 5, 227–231.
- Philpot J, Pennock J, Cowan F, et al. (2000) Brain magnetic resonance imaging abnormalities in merosin-positive congenital muscular dystrophy. Europ J Paediatr Neurol 4:109–14.
- Pini A, Merlini L, Tome FMS et al. (1996) Merosin negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia. Report from three Italian cases in two families. Brain Dev 18, 316–322.
- Rudnick-Schoneborn, Forkert R, Hahnen E et al. (1996) Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN deletion findings. Neuropediatrics 27, 8–15.
- Rutherford MA, Heckmatt JZ and Dubowitz V (1989) Congenital myotonic dystrophy: respiratory function at birth determines survival. Arch Dis Child 64, 191–195.
- Santavuori P, Somer H, Saino K et al. (1989) Muscle–eye–brain disease (MEB). Brain Dev 11, 147–153.
- Shorer Z, Philpot J, Muntoni F et al. (1995) Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J Child Neurol 10, 472–475.
- Sunada Y, Edgar TS, Lotz BP et al. (1995) Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology 45, 2084–2089.
- Takashima S and Mizuguchi M (1997) Cytoarchitectonic alterations of the cerebral cortex in Fukuyama type congenital muscular dystrophy and other cortical dysplasia syndrome. In: Fukuyama Y, Osawa M, and Saito K (Eds) Congenital Muscular Dystrophies. Amsterdam, Elsevier Science, pp. 125–142.
- Talim B, Ferreiro A, Cormand B, et al. (2000). Merosin-deficient congenital muscular dystrophy with mental retardation and cerebellar cysts unlinked to the LAMA2, FCMD and MEB loci. Neuromusc Disord 10, 548–52.
- Tanabe Y, Iai M, Tamai K et al. (1992) Neuroradiological findings in children with congenital myotonic dystrophy. Acta Paediatr 81, 613–617.
- Topaloglu H, Talim B, Vignier N, et al. (1998) Merosin-deficient congenital muscular dystrophy with severe mental retardation and normal cranial MRI: a report of two siblings. Neuromusc Disord 8, 169–174.
- Trevisan CP, Martinello F, Ferruzza E et al. (1996) Brain alteration in the classical form of congenital muscular dystrophy. Clinical and neuroimaging follow up of 12 cases and correlation with the expression of merosin in muscle. Child Nerv Syst 12, 604–610.
- Trevisan CP, Martinello F, Armani M et al. (1997) Brain involvement in a series of cases with merosin-positive congenital muscular dystrophy. Neuromusc Disord 7, 433.
- Tome FM, Evangelista T, Leclerc A et al. (1994) Congenital muscular dystrophy with merosin deficiency. CR Acad Sci III 317(4), 351–357.
- Villanova M, Malandrini A, Toti P et al. (1996) Localization of merosin in the normal human brain: implications for congenital muscular dystrophy with merosin deficiency. J Submicrosc Cytol Pathol 28(1), 1–4.
- Villanova M, Mercuri E, Bertini E, et al. (2001). Congenital muscular dystrophy associated with calf hyperthrophy, microcephaly and severe mental retardation: A new CMD syndrome. Neuromusc Disord 10: 541–547.
- Voit T (1998) Congenital muscular dystrophies: 1997 update. Brain Dev, 20 65–74.
- Vuolteenalho R, Nissinen M, Sainio K, et al. (1994). Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain human fetal tissues. J Cell Biol, 124:381–394.